R Packages for Communicating Reproducible Research
26 December 2019
Contents
Last modified: 31 May, 2019
1 Abstract
This tutorial is for all who wish to write R packages. R is a fantastic language for you to develop new statistical approaches for the analysis and comprehension of real-world data. R packages provide a way to capture your new approach in a reproducible, documented unit. An R package is surprisingly easy to create, and creating an R package has many benefits. In this tutorial we create an R package. We start with a data set and a simple script transforming the data in a useful way; perhaps you have your own data set and script? We replace the script with a function, and place the function and data into an R package. We then add documentation, so that our users (and our future selves) understand what the function does and how the function applies to new data sets. With an R package in hand, we can tackle more advance challenges: vignettes for rich narrative description of the package; unit tests to make our package more robust; and version control to document how we change the package. The final step in the development of our package is to share it with others, through github, through CRAN, or though domain-specific channels such as Bioconductor.
2 Biological and statistical motivation
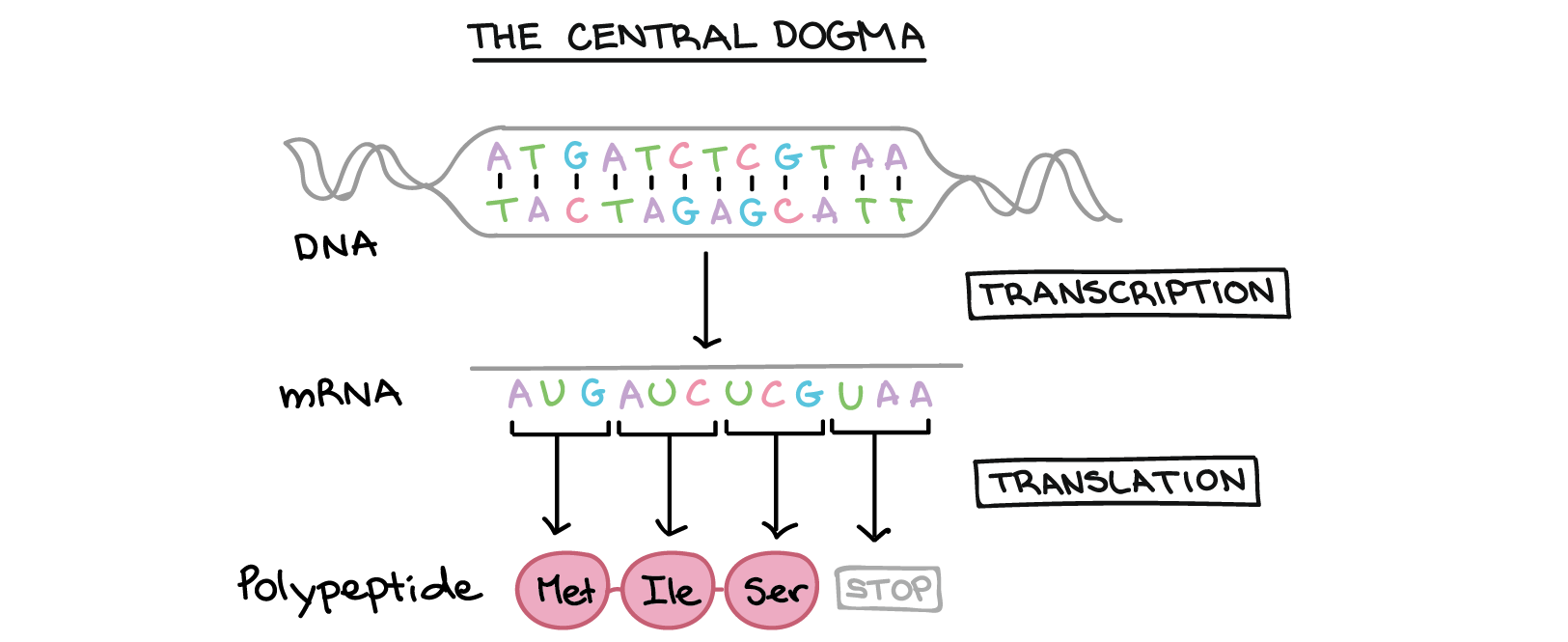
2.1 Measuring gene expression on single cells: single-cell RNA seq
The ‘central dogma’ of molecular biology: genes encoded in DNA (chromosomes) are transcribed to mRNA and then translated to protiens.

- https://cdn.kastatic.org/ka-perseus-images/2b597889d05bc601803a3b4d9ec5ccd5e7b8d3af.png All Khan Academy content is available for free at www.khanacademy.org
{kind=link}
RNA-sequencing (bulk RNA-seq)
- Isolate mRNA from a large sample of cells
- Reverse transcibe to cDNA, fragment, and sequence
- Align sequenced fragments to reference genome
- More fragments aligned interpretted as higher gene expression.

Single-cell RNA-seq
- Isolate individual cells
- Associate each cell with bar-coded beads
- Sequence bar-coded cDNA
- Most current methods lead to very sparse ‘coverage’

- Hwang et al., 2018 https://doi.org/10.1038/s12276-018-0071-8
2.2 Simulated data
Parameters:
n_genes <- 20000
n_cells <- 100
## gamma-distributed gene means
rate <- .1
shape <- .1
## negative binomial counts
dispersion <- 0.1A very rough simulation:
set.seed(123)
gene_means <- rgamma(n_genes, shape = shape, rate = rate)
cell_size_factors <- 2 ^ rnorm(n_cells, sd = 0.5)
cell_means <- outer(gene_means, cell_size_factors, `*`)
counts <- matrix(
rnbinom(n_genes * n_cells, mu = cell_means, size = 1 / dispersion),
nrow = n_genes, ncol = n_cells
)Basic properties of the simulated data
range(counts)
## [1] 0 256
## proportion of zeros
mean(counts == 0)
## [1] 0.793133
## 'library size' -- reads mapped per cell
hist(colSums(counts), main = "Library Size")
## average experssion per gene
hist(rowMeans(log1p(counts)), main = "log Gene Expression")
2.3 Size factors
log_counts <- log(counts)
centered <- log_counts - rowMeans(log_counts)
filtered_median <- function(x)
median(x[is.finite(x)])
size_factors <- exp(apply(centered, 2, filtered_median))
hist(size_factors)
range(size_factors)
## [1] 0.4438464 2.8639456
median(size_factors)
## [1] 1.0257813 The basics
3.1 R packages
Collection of files and directories on disk. A complete package might have a structure like that illustrated below.
SCSimulate
DESCRIPTION
NAMESPACE
R/
simulate.R
size_factors.R
man/
simulate.Rd
size_factors.Rd
vignettes/
Using_this_package.Rmd
tests/
testthat.R
testthat/
test_simulate.R
test_size_factor.RDESCRIPTION
- Package name, title, description (like paper abstract)
- Authors and maintainer (contact author)
- License
Other packages this package depends on (‘dependencies’)
Depends: Data structures or work flows required for use of this package.Imports: Used inside the current package. For instance, we will use function likergamma(),rnorm(), andrnbinom()from thestatspackage.Suggests: Used in examples or vignettes.
NAMESPACE
- Functions used by this package –
import(),importFrom() - Functions this package makes available to users –
export()
R/
- Text files containing R function definitions
man/
- Text files documenting functions
vignettes/
- ‘Markdown’ or other text documents describing use of the package.
tests/
- Functions used to provide ‘unit’ tests for the package.
3.2 Package skeleton
Create a package
devtools::create("SCSimulate")
## ✔ Creating 'SCSimulate/'
## ✔ Setting active project to '/Users/ma38727/b/github/BiocIntro/vignettes/SCSimulate'
## ✔ Creating 'R/'
## ✔ Writing 'DESCRIPTION'
## Package: SCSimulate
## Title: What the Package Does (One Line, Title Case)
## Version: 0.0.0.9000
## Authors@R (parsed):
## * First Last <[email protected]> [aut, cre] (YOUR-ORCID-ID)
## Description: What the package does (one paragraph).
## License: What license it uses
## Encoding: UTF-8
## LazyData: true
## ✔ Writing 'NAMESPACE'
## ✔ Setting active project to '<no active project>'Edit the DESCRIPTION file (plain text)
Package: SCSimulate
Title: Simulate single-cell RNA seq data
Version: 0.0.0.9000
Authors@R:
c(person(
given = "Martin",
family = "Morgan",
role = c("aut", "cre"),
email = "[email protected]",
comment = c(ORCID = "YOUR-ORCID-ID")
), person(
given = "Another",
family = "Author",
role = "aut"
))
Description: This package simulates single-cell RNA seq data using a gamma
distribution of gene expression values, and negative binomial model of
counts per cell. The package also contains functions for preprocessing,
including a simple calculation of library scaling factors.
License: Artistic-2.0
Imports: stats
Encoding: UTF-8
LazyData: trueSo far, our package looks like
SCSimulate
DESCRIPTION
NAMESPACE
R/3.3 From script to function
Transform the part of the script describing the simulation to a
function simulate(). Use function arguments to capture default
values.
simulate <-
function(n_genes = 20000, n_cells = 100,
rate = 0.1, shape = 0.1, dispersion = 0.1)
{
gene_means <- rgamma(n_genes, shape = shape, rate = rate)
cell_size_factors <- 2 ^ rnorm(n_cells, sd = 0.5)
cell_means <- outer(gene_means, cell_size_factors, `*`)
matrix(
rnbinom(n_genes * n_cells, mu = cell_means, size = 1 / dispersion),
nrow = n_genes, ncol = n_cells
)
}Transform the part of the script describing calculation of size
factors to a function size_factors(). The only argument to
size_factors() is a matrix of counts.
.filtered_median <- function(x)
median(x[is.finite(x)])
size_factors <-
function(counts)
{
log_counts <- log(counts)
centered <- log_counts - rowMeans(log_counts)
exp(apply(centered, 2, .filtered_median))
}Check that we haven’t made any mistakes.
set.seed(123)
counts <- simulate()
size_factors <- size_factors(counts)
range(size_factors)
## [1] 0.4438464 2.8639456
median(size_factors)
## [1] 1.0257813.4 Add function definitions to the package
Place functions into files in the R/ directory. Typically, name the
file after the function / group of functions in the file. E.g.,
file: R/simulate.R
simulate <-
function(n_genes = 20000, n_cells = 100,
rate = 0.1, shape = 0.1, dispersion = 0.1)
{
gene_means <- rgamma(n_genes, shape = shape, rate = rate)
cell_size_factors <- 2 ^ rnorm(n_cells, sd = 0.5)
cell_means <- outer(gene_means, cell_size_factors, `*`)
matrix(
rnbinom(n_genes * n_cells, mu = cell_means, size = 1 / dispersion),
nrow = n_genes, ncol = n_cells
)
}file: R/size_factors.R
.filtered_median <- function(x)
median(x[is.finite(x)])
size_factors <-
function(counts)
{
log_counts <- log(counts)
centered <- log_counts - rowMeans(log_counts)
exp(apply(centered, 2, .filtered_median))
}Our package now looks like
SCSimulate
DESCRIPTION
NAMESPACE
R/
simulate.R
size_factors.R3.5 Document the functions
Use roxygen2 for documentation by adding tagged lines starting with
#' immediatly above each function. Common tags are illustrated
below.
@titleis a one-line description appearing at the top of a help page.@descriptionprovides a short description of the function, presented after the title. Use@detailsfor more extensive description appearing after the ‘Usage’ section (generated based on the signature of the function after the@exporttag) of a help page.- Document parameter (
@param) and return (@return) values carefully. The@paramvalues are used to form the ‘Arguments’ section of the help page. The@returnvalue appears in the ‘Returns’ section of the help page. @examplesare include in the ‘Examples’ section of the help page, and must be complete and syntactically correct R code (examples are evaluated when a package is built and checked).- Use
@importFromto indicate that a particular package provides specific functions used in the current package. - For readability, ‘wrapped’ lines to 80 columns. Use indentation and spacing consistently and generously.
file: R/simulate.R
#' @title Simulate single-cell data
#'
#' @description `simulate()` produces a genes x cells count matrix of
#' simulated single-cell RNA-seq data. Gene expression is modelled
#' using a gamma distribution. Counts are simulated using a
#' negative binomial distribution.
#'
#' @param n_genes integer(1) the number of genes (rows) to simulate.
#'
#' @param n_cells integer(1) the number of cells (columns) to simulate.
#'
#' @param rate numeric(1) rate parameter of the `rgamma()` distribution.
#'
#' @param shape numeric(1) shape parameter of the `rgamma()` distribution.
#'
#' @param dispersion numeric(1) size (`1 / dispersion`) parameter of
#' the `rnbinom()` distribution.
#'
#' @return `simulate() returns a `n_genes x n_cells` matrix of
#' simulated single-cell RNA-seq counts.
#'
#' @examples
#' counts <- simulate()
#' dim(counts)
#' mean(counts == 0) # fraction of '0' cells
#' range(counts)
#'
#' @importFrom stats rgamma rnorm rnbinom
#'
#' @export
simulate <-
function(n_genes = 20000, n_cells = 100,
rate = 0.1, shape = 0.1, dispersion = 0.1)
{
gene_means <- rgamma(n_genes, shape = shape, rate = rate)
cell_size_factors <- 2 ^ rnorm(n_cells, sd = 0.5)
cell_means <- outer(gene_means, cell_size_factors, `*`)
matrix(
rnbinom(n_genes * n_cells, mu = cell_means, size = 1 / dispersion),
nrow = n_genes, ncol = n_cells
)
}file: R/size_factors.R
#' @importFrom stats median
.filtered_median <- function(x)
median(x[is.finite(x)])
#' @title Calculate geometric mean-centered median scaled cell scaling
#' factors.
#'
#' @description `size_factors()` centers the log counts of each row
#' (gene) by the row mean of the log counts. The finite centered
#' values are then used to compute column-wise geometric median
#' scaling factors.
#'
#' @param counts matrix() of gene x cell RNA-seq counts.
#'
#' @return `size_factors()` returns a `numeric(ncol(counts))` vector
#' of scaling factors.
#'
#' @examples
#' set.seed(123)
#' counts <- simulate()
#' size_factors <- size_factors(counts)
#' median(size_factors) # approximately 1
#'
#' @export
size_factors <-
function(counts)
{
log_counts <- log(counts)
centered <- log_counts - rowMeans(log_counts)
exp(apply(centered, 2, .filtered_median))
}3.6 Update ‘NAMESPACE’ and ‘man’ pages
devtools::document("SCSimulate")
## Updating SCSimulate documentation
## Writing NAMESPACE
## Loading SCSimulate
## Writing NAMESPACEUpdates the NAMESPACE file
- Functions used by this package (
stats::rgamma(),stats::rnorm(),stats::rnbinom()). - Indicates functions defined by this package and meant to be
visible to the user (
simulate()andsize_factors(), but not.filtered_median()).
- Functions used by this package (
Transforms the documentation introduced above into stand-alone files
- E.g.,
man/simulate.Rd - A plain text file, but with LaTeX-style markup understood by R.
- E.g.,
Our package now looks like
SCSimulate
DESCRIPTION
NAMESPACE
R/
simulate.R
size_factors.R
man/
simulate.Rd
size_factors.RdThe NAMESPACE file has been updated to
cat(readLines("SCSimulate/NAMESPACE"), sep="\n")
## # Generated by roxygen2: do not edit by hand
##
## export(simulate)
## export(size_factors)
## importFrom(stats,median)
## importFrom(stats,rgamma)
## importFrom(stats,rnbinom)
## importFrom(stats,rnorm)3.7 Build & check
- Build (collate) package files into a ‘tar’ ball,
SCSimulate_0.0.0.9000.tar.gz. - Check that the tar ball is complete and correct.
devtools::check("SCSimulate")
## Updating SCSimulate documentation
## Writing NAMESPACE
## Loading SCSimulate
## Writing NAMESPACE
## Writing size_factors.Rd
## ── Building ────────────────────────────────────────────────────── SCSimulate ──
## Setting env vars:
## ● CFLAGS : -Wall -pedantic -fdiagnostics-color=always
## ● CXXFLAGS : -Wall -pedantic -fdiagnostics-color=always
## ● CXX11FLAGS: -Wall -pedantic -fdiagnostics-color=always
## ────────────────────────────────────────────────────────────────────────────────
## ✔ checking for file ‘/Users/ma38727/a/github/BiocIntro/vignettes/SCSimulate/DESCRIPTION’
## ─ preparing ‘SCSimulate’:
## ✔ checking DESCRIPTION meta-information
## ─ checking for LF line-endings in source and make files and shell scripts
## ─ checking for empty or unneeded directories
## ─ building ‘SCSimulate_0.0.0.9000.tar.gz’
##
## ── Checking ────────────────────────────────────────────────────── SCSimulate ──
## Setting env vars:
## ● _R_CHECK_CRAN_INCOMING_USE_ASPELL_: TRUE
## ● _R_CHECK_CRAN_INCOMING_REMOTE_ : FALSE
## ● _R_CHECK_CRAN_INCOMING_ : FALSE
## ● _R_CHECK_FORCE_SUGGESTS_ : FALSE
## ── R CMD check ─────────────────────────────────────────────────────────────────
## Bioconductor version 3.11 (BiocManager 1.30.10), ?BiocManager::install for help
## ─ using log directory '/private/var/folders/yn/gmsh_22s2c55v816r6d51fx1tnyl61/T/Rtmp6S4exQ/SCSimulate.Rcheck'
## ─ using R Under development (unstable) (2019-12-01 r77489)
## ─ using platform: x86_64-apple-darwin17.7.0 (64-bit)
## ─ using session charset: UTF-8
## ─ using options '--no-manual --as-cran'
## ✔ checking for file 'SCSimulate/DESCRIPTION'
## ─ this is package 'SCSimulate' version '0.0.0.9000'
## ─ package encoding: UTF-8
## ✔ checking package namespace information
## ✔ checking package dependencies (3.4s)
## ✔ checking if this is a source package
## ✔ checking if there is a namespace
## ✔ checking for executable files
## ✔ checking for hidden files and directories
## ✔ checking for portable file names
## ✔ checking for sufficient/correct file permissions
## ✔ checking serialization versions
## ✔ checking whether package 'SCSimulate' can be installed (1.8s)
## ✔ checking installed package size
## ✔ checking package directory
## ✔ checking for future file timestamps (505ms)
## ✔ checking DESCRIPTION meta-information
## ✔ checking top-level files
## ✔ checking for left-over files
## ✔ checking index information
## ✔ checking package subdirectories
## ✔ checking R files for non-ASCII characters
## ✔ checking R files for syntax errors
## ✔ checking whether the package can be loaded
## ✔ checking whether the package can be loaded with stated dependencies
## ✔ checking whether the package can be unloaded cleanly
## ✔ checking whether the namespace can be loaded with stated dependencies
## ✔ checking whether the namespace can be unloaded cleanly
## ✔ checking loading without being on the library search path
## ✔ checking dependencies in R code
## ✔ checking S3 generic/method consistency (651ms)
## ✔ checking replacement functions
## ✔ checking foreign function calls
## ✔ checking R code for possible problems (2.1s)
## ✔ checking Rd files
## ✔ checking Rd metadata
## ✔ checking Rd line widths
## ✔ checking Rd cross-references
## ✔ checking for missing documentation entries
## ✔ checking for code/documentation mismatches (407ms)
## ✔ checking Rd \usage sections (792ms)
## ✔ checking Rd contents
## ✔ checking for unstated dependencies in examples
## ✔ checking examples (1.7s)
## ✔ checking for non-standard things in the check directory
## ✔ checking for detritus in the temp directory
##
##
## ── R CMD check results ────────────────────────────── SCSimulate 0.0.0.9000 ────
## Duration: 14.9s
##
## 0 errors ✔ | 0 warnings ✔ | 0 notes ✔3.8 Install
devtools::install("SCSimulate")
## ✔ checking for file ‘/Users/ma38727/a/github/BiocIntro/vignettes/SCSimulate/DESCRIPTION’
## ─ preparing ‘SCSimulate’:
## ✔ checking DESCRIPTION meta-information
## ─ checking for LF line-endings in source and make files and shell scripts
## ─ checking for empty or unneeded directories
## ─ building ‘SCSimulate_0.0.0.9000.tar.gz’
##
## Running /Users/ma38727/bin/R-devel/bin/R CMD INSTALL \
## /var/folders/yn/gmsh_22s2c55v816r6d51fx1tnyl61/T//Rtmp6S4exQ/SCSimulate_0.0.0.9000.tar.gz \
## --install-tests
## * installing to library ‘/Users/ma38727/Library/R/4.0/Bioc/3.11/library’
## * installing *source* package ‘SCSimulate’ ...
## ** using staged installation
## ** R
## ** byte-compile and prepare package for lazy loading
## ** help
## *** installing help indices
## ** building package indices
## ** testing if installed package can be loaded from temporary location
## ** testing if installed package can be loaded from final location
## ** testing if installed package keeps a record of temporary installation path
## * DONE (SCSimulate)3.9 Use!
library(SCSimulate)?simulate
?size_factorsexample(size_factors)
##
## sz_fct> set.seed(123)
##
## sz_fct> counts <- simulate()
##
## sz_fct> size_factors <- size_factors(counts)
##
## sz_fct> median(size_factors) # approximately 1
## [1] 1.0257814 Advanced challenges
4.1 Data sets
4.2 Communicate: vignettes
4.3 Robust: tests & examples
4.4 Mature: version control
4.5 Dissemination
5 sessionInfo()
sessionInfo()
## R Under development (unstable) (2019-12-01 r77489)
## Platform: x86_64-apple-darwin17.7.0 (64-bit)
## Running under: macOS High Sierra 10.13.6
##
## Matrix products: default
## BLAS: /Users/ma38727/bin/R-devel/lib/libRblas.dylib
## LAPACK: /Users/ma38727/bin/R-devel/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] SCSimulate_0.0.0.9000 BiocStyle_2.15.2
##
## loaded via a namespace (and not attached):
## [1] Rcpp_1.0.3 compiler_4.0.0 BiocManager_1.30.10
## [4] prettyunits_1.0.2 remotes_2.1.0 tools_4.0.0
## [7] testthat_2.3.1 digest_0.6.23 pkgbuild_1.0.6
## [10] pkgload_1.0.2 evaluate_0.14 memoise_1.1.0
## [13] rlang_0.4.2 rstudioapi_0.10 cli_2.0.0
## [16] yaml_2.2.0 xopen_1.0.0 xfun_0.11
## [19] xml2_1.2.2 roxygen2_7.0.2 withr_2.1.2
## [22] stringr_1.4.0 knitr_1.26 desc_1.2.0
## [25] fs_1.3.1 devtools_2.2.1 rprojroot_1.3-2
## [28] glue_1.3.1 R6_2.4.1 processx_3.4.1
## [31] fansi_0.4.0 rcmdcheck_1.3.3 rmarkdown_2.0
## [34] bookdown_0.16 sessioninfo_1.1.1 purrr_0.3.3
## [37] whisker_0.4 callr_3.4.0 magrittr_1.5
## [40] codetools_0.2-16 clisymbols_1.2.0 backports_1.1.5
## [43] ps_1.3.0 ellipsis_0.3.0 htmltools_0.4.0
## [46] usethis_1.5.1 assertthat_0.2.1 stringi_1.4.3
## [49] crayon_1.3.4