---
title: "Introduction to smokingMouse"
author:
- name: Daianna Gonzalez-Padilla
affiliation:
- Lieber Institute for Brain Development (LIBD)
email: glezdaianna@gmail.com
output:
BiocStyle::html_document:
self_contained: yes
toc: true
toc_float: true
toc_depth: 2
code_folding: show
date: "`r doc_date()`"
package: "`r pkg_ver('smokingMouse')`"
vignette: >
%\VignetteIndexEntry{Introduction to smokingMouse}
%\VignetteEngine{knitr::rmarkdown}
%\VignetteEncoding{UTF-8}
---
```{r, include = FALSE}
knitr::opts_chunk$set(
collapse = TRUE,
comment = "#>"
)
```
```{r vignetteSetup, echo=FALSE, message=FALSE, warning = FALSE}
## Track time spent on making the vignette
startTime <- Sys.time()
## Bib setup
library("RefManageR")
## Write bibliography information
bib <- c(
R = citation(),
AnnotationHubData = citation("AnnotationHubData")[1],
ExperimentHub = citation("ExperimentHub")[1],
BiocStyle = citation("BiocStyle")[1],
knitr = citation("knitr")[1],
RefManageR = citation("RefManageR")[1],
rmarkdown = citation("rmarkdown")[1],
sessioninfo = citation("sessioninfo")[1],
testthat = citation("testthat")[1]
)
```
# Introduction
Welcome to the `smokingMouse` project.
In this vignette we'll show you how to access the smokingMouse LIBD datasets `r Citep(bib[['smokingMouse']])`.
You can find the analysis code and the data generation in [here](https://github.com/LieberInstitute/smokingMouse_Indirects).
## Motivation
The main motivation to create this bioconductor package was to provide public and free access to all RNA-seq datasets that were generated for the smokingMouse project, containing many variables of interest that make it possible to answer a wide range of biological questions related to smoking and nicotine effects in mice.
## Overview
This bulk RNA-sequencing project consisted of a differential expression analysis (DEA) involving 4 data types: genes, exons, transcripts and junctions. The main goal of this study was to explore the effects of prenatal exposure to maternal smoking and nicotine exposures on the developing mouse brain. As secondary objectives, this work evaluated: 1) the affected genes by each exposure on the adult female brain in order to compare offspring and adult results and 2) the effects of smoking on adult blood and brain to search for overlapping biomarkers in both tissues. Finally, DEGs identified in mice were compared against previously published results from a human study (Semick, S.A. et al. (2018)).
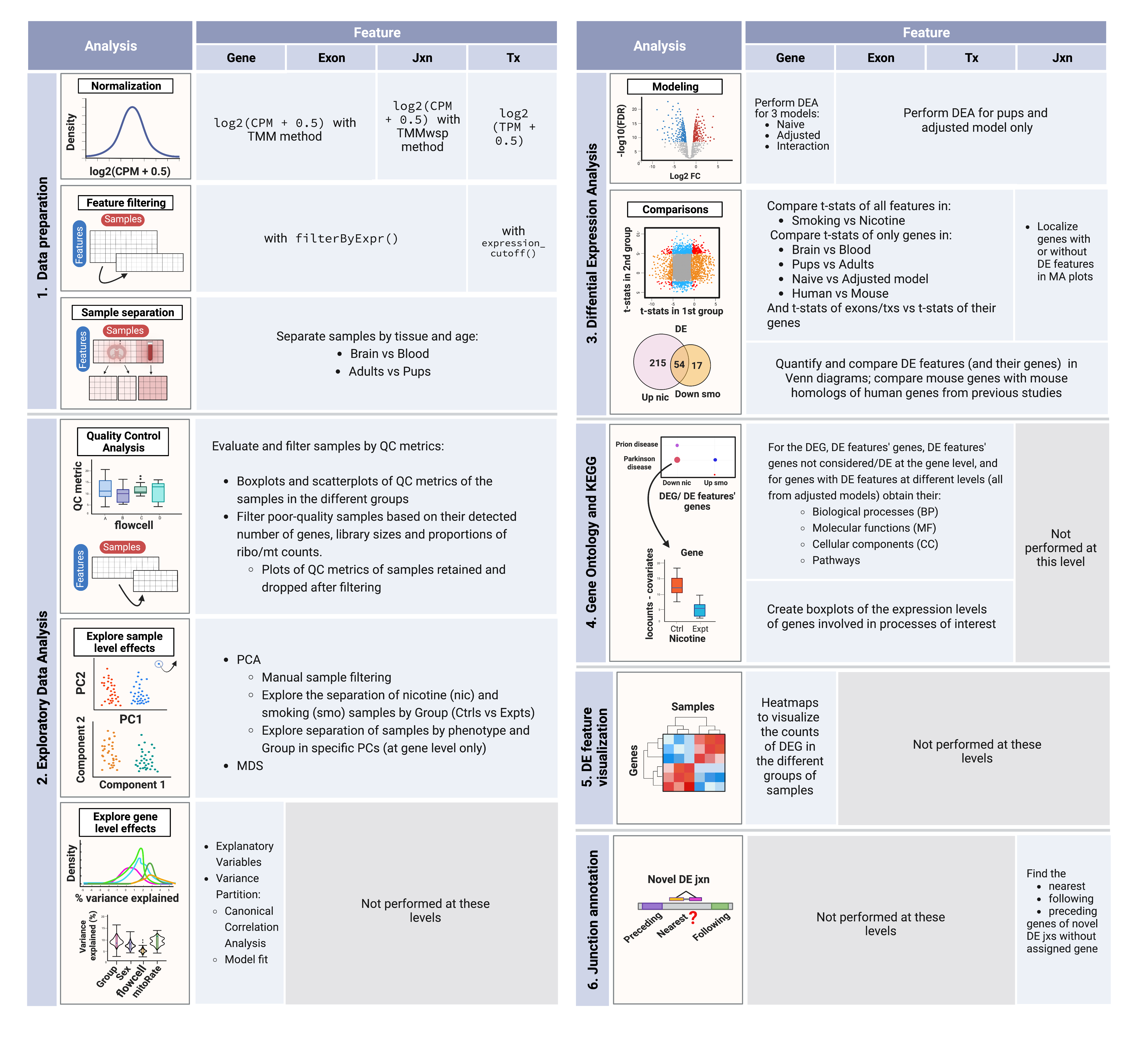
The next table summarizes the analyses done at each level.
Table 1: 1. Data preparation: in this first step, counts of genes, exons and junctions were normalized to CPM and scaled; transcript expression values were only scaled since they were already in TMP. Then, low-expression features were removed using the indicated methods and samples were separated by tissue and age in order to create subsets of the data for downstream analyses. 2. Exploratory Data Analysis: QC metrics of the samples were examined and used to filter them; sample level effects were explored through dimensionality reduction methods and rare samples in PCA plots were manually removed from the datasets; gene level effects were evaluated with analyses of explanatory variables and variance partition. 3. Differential Expression Analysis: with the relevant variables identified in the previous steps, the DEA was performed at the gene level for nicotine and smoking, adult and pup, and blood and brain samples, and for 3 models: the naive one modeled ~Group + batch effects, the adjusted model modeled ~Group + Pregnancy + batch effects for adults and ~Group + Sex + batch effects for pups, and the interaction model ~Group\*Pregnancy + batch effects for adults and ~Group*Sex + batch effects for pups; DEA on the rest of the levels was performed for pups only and using the adjusted model. After that, signals of the features in nicotine and smoking were compared, as well as the signals of exons and txs vs the effects of their genes, and genes’ signals were additionally compared in the different tissues, ages, models and species (vs human data of a previous study). All resultant DEG and DE features (and their genes) were quantified and compared based on their experiment (nic/smo) and direction of regulation (up/down); DEG were further compared against genes of DE exons and txs; mouse genes were also compared with human genes affected by cigarette smoke or associated with TUD. 4. Gene Ontology and KEGG: taking the DEG and the genes of DE txs and exons, GO & KEGG analyses were done and the expression levels of genes that participate in brain development related processes were explored. 5. DE feature visualization: DEG counts were represented in heatmaps in order to distinguish the groups of up and down-regulated genes. 6. Junction annotation: for novel DE jxns of unknown gene, their nearest, preceding and following genes were determined.
Abbreviations: Jxn: junction; Tx: transcript; CPM: counts per million; TPM: transcripts per million; TMM: Trimmed Mean of M-Values; TMMwsp: TMM with singleton pairing; EDA: exploratory data analysis; QC: quality control; ribo: ribosomal; mt: mitochondrial; PCA: Principal Component Analysis; PC: principal component; MDS: Multidimensional Scaling; DEA: differential expression analysis; DE: differential expression/differentially expressed; DEG: differentially expressed genes; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; TUD: tobacco use disorder.
## Study design
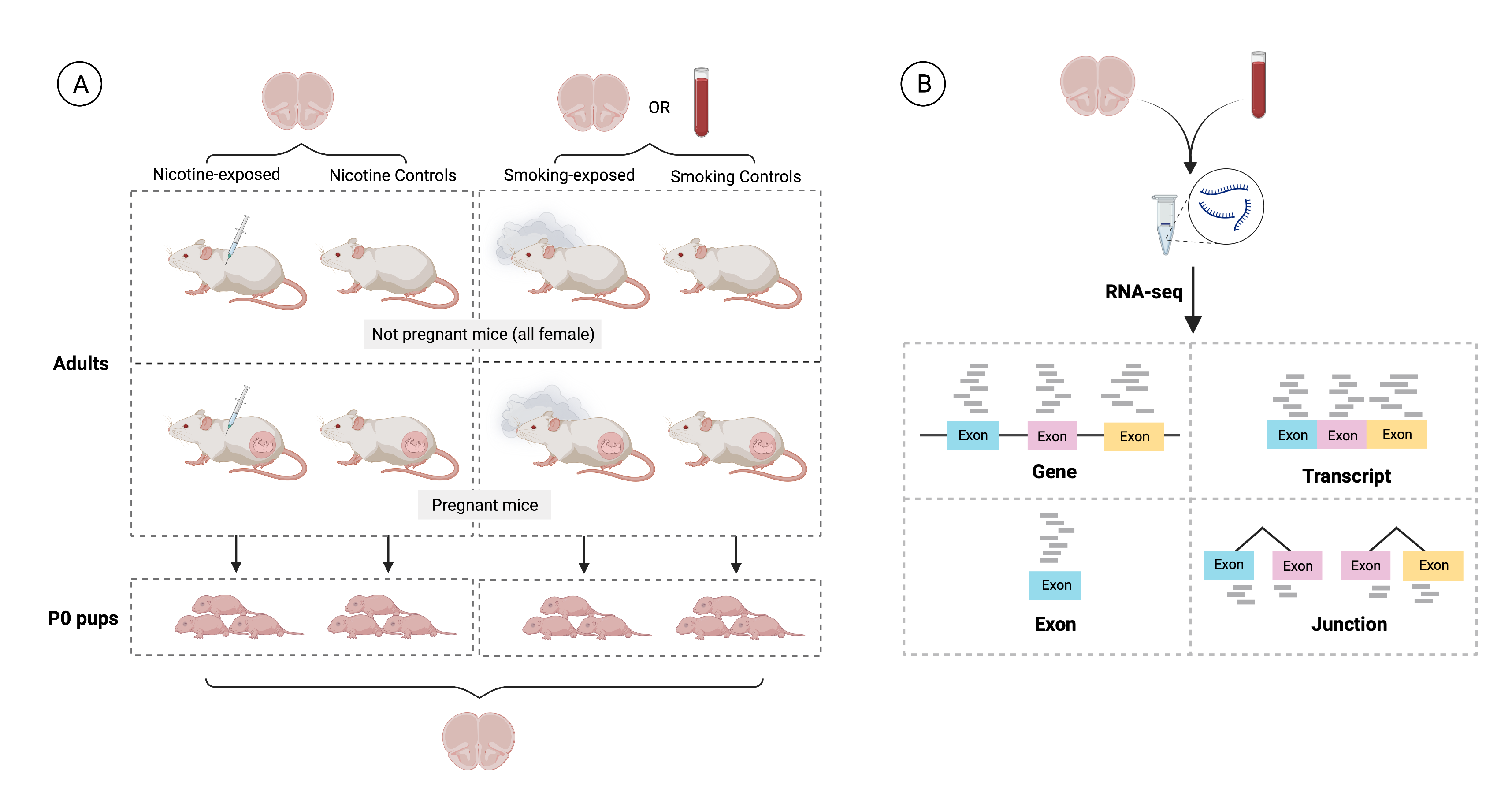
Figure 1: Experimental design of the study. A) 36 pregnant dams and 35 non-pregnant female adult mice were either administered nicotine by intraperitoneal injection (IP; n=12), exposed to cigarette smoke in smoking chambers (n=24), or controls (n=35; 11 nicotine controls and 24 smoking controls). A total of 137 pups were born to pregnant dams: 19 were born to mice that were administered nicotine, 46 to mice exposed to cigarette smoke and the remaining 72 to control mice (23 to nicotine controls and 49 to smoking controls). Samples from frontal cortices of P0 pups and adults were obtained, as well as blood samples from smoking-exposed and smoking control adults. B) RNA was extracted, RNA-seq libraries were prepared and sequenced to obtain expression counts for genes, exons, transcripts and exon-exon junctions.
# Basics
## Install `smokingMouse`
`R` is an open-source statistical environment which can be easily modified to enhance its functionality via packages.
`r Biocpkg("smokingMouse")` is a `R` package available via the [Bioconductor](http://bioconductor.org) repository for packages.
`R` can be installed on any operating system from [CRAN](https://cran.r-project.org/) after which you can install `r Biocpkg("smokingMouse")` by using the following commands in your `R` session:
```{r "install", eval = FALSE}
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("smokingMouse")
## Check that you have a valid Bioconductor installation
BiocManager::valid()
```
## Required knowledge
`r Biocpkg("smokingMouse")` is based on many other packages and in particular in those that have implemented the infrastructure needed for dealing with RNA-seq data and differential expression.
That is, packages like `r Biocpkg("SummarizedExperiment")` and `r Biocpkg("limma")`.
If you are asking yourself the question "Where do I start using Bioconductor?" you might be interested in [this blog post](http://lcolladotor.github.io/2014/10/16/startbioc/#.VkOKbq6rRuU).
## Asking for help
As package developers, we try to explain clearly how to use our packages and in which order to use the functions. But `R` and `Bioconductor` have a steep learning curve so it is critical to learn where to ask for help.
The blog post quoted above mentions some but we would like to highlight the [Bioconductor support site](https://support.bioconductor.org/) as the main resource for getting help: remember to use the `smokingMouse` tag and check [the older posts](https://support.bioconductor.org/tag/smokingMouse/).
Other alternatives are available such as creating GitHub issues and tweeting. However, please note that if you want to receive help you should adhere to the [posting guidelines](http://www.bioconductor.org/help/support/posting-guide/).
It is particularly critical that you provide a small reproducible example and your session information so package developers can track down the source of the error.
## Citing `smokingMouse`
We hope that `r Biocpkg("smokingMouse")` will be useful for your research. Please use the following information to cite the package and the overall approach.
```{r "citation"}
## Citation info
citation("smokingMouse")
```
# Quick start to using `smokingMouse`
To get started, please load the `r Biocpkg('smokingMouse')` package.
```{r "start", message=FALSE}
library("smokingMouse")
```
# smoking Mouse datasets
The raw data was generated by LIBD researchers and is composed of expression counts of genes, transcripts (txs), exons and exon-exon junctions (jxns) across 208 mice samples (brain/blood; adult/pup; nicotine-exposed/smoking-exposed/controls).
The datasets available in `r Biocpkg("smokingMouse")` were generated by Daianna Gonzalez-Padilla.
The human data was generated by Semick, S.A. et al. (2018) in Mol Psychiatry, DOI: https://doi.org/10.1038/s41380-018-0223-1 and it contains the results of a DEA in adult and prenatal human brain samples exposed to cigarette smoke.
## Description of the datasets
### Mouse datasets:
* They are 4 `r Biocpkg('RangedSummarizedExperiment')` (RSE) objects that contain feature info in `rowData(RSE)` and sample info in `colData(RSE)`.
* Raw expression counts can be accessed with `assays(RSE)$counts` and the lognorm counts (log2(cpm + 0.5) for genes, exons and jxns; log2(tpm + 0.5) for txs) with `assays(RSE)$logcounts`.
### Human datasets:
* They are two data frames with the information of human genes and some relevant statistical metrics of differential expression (DE).
## Data specifics
* *'rse_gene_mouse_RNAseq_nic-smo.Rdata'*: (rse_gene object) the gene RSE object contains raw and normalized expression data of 55401 mouse genes across 208 samples from brains and blood of healthy and nicotine/smoking-exposed pup and adult mice.
* *'rse_tx_mouse_RNAseq_nic-smo.Rdata'*: (rse_tx object) the tx RSE object contains raw and normalized expression data of 142604 mouse transcripts across 208 samples from brains and blood of healthy and nicotine/smoking-exposed pup and adult mice.
* *'rse_exon_mouse_RNAseq_nic-smo.Rdata'*: (rse_exon object) the exon RSE object contains raw and normalized expression data of 447670 mouse exons across 208 samples from brains and blood of healthy and nicotine/smoking-exposed pup and adult mice.
* *'rse_jx_mouse_RNAseq_nic-smo.Rdata'*: (rse_jx object) the jx RSE object contains raw and normalized expression data of 1436068 mouse exon-exon junctions across 208 samples from brains and blood of healthy and nicotine/smoking-exposed pup and adult mice.
All the above datasets contain sample and feature information and additional data of the results obtained in the filtering steps and the DEA.
* *'de_genes_prenatal_human_brain_smoking.Rdata'*: (object with the same name) data frame with DE (ctrls vs smoking-exposed samples) data of 18067 genes in human prenatal brain samples exposed to cigarette smoke.
* *'de_genes_adult_human_brain_smoking.Rdata'*: (object with the same name) data frame with DE (ctrls vs smoking-exposed samples) data of 18067 genes in human adult brain samples exposed to cigarette smoke.
## Variables of mice data
Feature information in `rowData(RSE)` contains the following variables:
* `retained_after_feature_filtering`: Boolean variable that equals TRUE if the feature passed the feature filtering based on expression levels and FALSE if not. Check code in [here](https://github.com/LieberInstitute/smokingMouse_Indirects/blob/main/code/02_build_objects/02_build_objects.R).
* `DE_in_adult_brain_nicotine`: Boolean variable that equals TRUE if the feature is differentially expressed (DE) in adult brain samples exposed to nicotine and FALSE if not. Check code in [here](https://github.com/LieberInstitute/smokingMouse_Indirects/tree/main/code/04_DEA).
* `DE_in_adult_brain_smoking`: Boolean variable that equals TRUE if the feature is differentially expressed (DE) in adult brain samples exposed to cigarette smoke and FALSE if not. Check code in [here](https://github.com/LieberInstitute/smokingMouse_Indirects/tree/main/code/04_DEA).
* `DE_in_adult_blood_smoking`: Boolean variable that equals TRUE if the feature is differentially expressed (DE) in adult blood samples exposed to cigarette smoke and FALSE if not. Check code in [here](https://github.com/LieberInstitute/smokingMouse_Indirects/tree/main/code/04_DEA).
* `DE_in_pup_brain_nicotine`: Boolean variable that equals TRUE if the feature is differentially expressed (DE) in pup brain samples exposed to nicotine and FALSE if not. Check code in [here](https://github.com/LieberInstitute/smokingMouse_Indirects/tree/main/code/04_DEA).
* `DE_in_pup_brain_smoking`: Boolean variable that equals TRUE if the feature is differentially expressed (DE) in pup brain samples exposed to cigarette smoke and FALSE if not. Check code in [here](https://github.com/LieberInstitute/smokingMouse_Indirects/tree/main/code/04_DEA).
The rest of the variables are outputs of SPEAQeasy pipeline. See [here](http://research.libd.org/SPEAQeasy/outputs.html) for a description of them.
Sample information in `colData(RSE)` contains the following variables:
* The Quality Control (QC) variables `sum`,`detected`,`subsets_Mito_sum`, `subsets_Mito_detected`, `subsets_Mito_percent`, `subsets_Ribo_sum`,`subsets_Ribo_detected` and `subsets_Ribo_percent` are returned by `addPerCellQC()`. See [here](https://rdrr.io/bioc/scuttle/man/addPerCellQC.html) for more info.
* `retained_after_QC_sample_filtering`: Boolean variable that equals TRUE if the sample passed the sample filtering based on QC metrics and FALSE if not. Check code in [here](https://github.com/LieberInstitute/smokingMouse_Indirects/blob/main/code/03_EDA/02_QC.R).
* `retained_after_manual_sample_filtering`: Boolean variable that equals TRUE if the sample passed the manual sample filtering based on PCA plots and FALSE if not. Check code in [here](https://github.com/LieberInstitute/smokingMouse_Indirects/blob/main/code/03_EDA/03_PCA_MDS.R)
The rest of the variables are outputs of SPEAQeasy. See [here](http://research.libd.org/SPEAQeasy/outputs.html) for their description.
## Variables of human data
Check [here](https://github.com/LieberInstitute/Smoking_DLPFC_Devel) to see the data generation and variables meaning.
## Downloading the data with `smokingMouse`
Using `r Biocpkg('smokingMouse')` `r Citep(bib[['smokingMouse']])` you can download these R objects. They are hosted by [Bioconductor](http://bioconductor.org/)'s `r Biocpkg('ExperimentHub')` `r Citep(bib[['ExperimentHub']])` resource.
Below you can see how to obtain these objects.
```{r 'experiment_hub'}
## Load ExperimentHub for downloading the data
library("ExperimentHub")
## Connect to ExperimentHub
ehub <- ExperimentHub::ExperimentHub()
## Load the datasets of the package
myfiles <- query(ehub, "smokingMouse")
## Resulting smokingMouse files from our ExperimentHub query
myfiles
```
```{r 'download_data'}
## Load SummarizedExperiment which defines the class container for the data
library("SummarizedExperiment")
######################
# Mouse data
######################
myfiles["EH8313"]
## Download the mouse gene data
# EH8313 | rse_gene_mouse_RNAseq_nic-smo
rse_gene <- myfiles[["EH8313"]]
## This is a RangedSummarizedExperiment object
rse_gene
## Optionally check the memory size
# lobstr::obj_size(rse_gene)
# 159.68 MB
## Check sample info
head(colData(rse_gene), 3)
## Check gene info
head(rowData(rse_gene), 3)
## Access the original counts
class(assays(rse_gene)$counts)
## Access the log normalized counts
class(assays(rse_gene)$logcounts)
dim(assays(rse_gene)$logcounts)
assays(rse_gene)$logcounts[1:3, 1:3]
######################
# Human data
######################
myfiles["EH8318"]
## Download the human gene data
# EH8318 | de_genes_adult_human_brain_smoking
de_genes_prenatal_human_brain_smoking <- myfiles[["EH8318"]]
## This is a GRanges object
class(de_genes_prenatal_human_brain_smoking)
de_genes_prenatal_human_brain_smoking
## Optionally check the memory size
# lobstr::obj_size(de_genes_prenatal_human_brain_smoking)
# 3.73 MB
## Access data of human genes as normally do with other GenomicRanges::GRanges()
## objects or re-cast it as a data.frame
de_genes_df <- as.data.frame(de_genes_prenatal_human_brain_smoking)
head(de_genes_df)
```
# Reproducibility
The `r Biocpkg("smokingMouse")` package `r Citep(bib[["smokingMouse"]])` and the [smoking mouse](https://github.com/LieberInstitute/smokingMouse_Indirects) project were made possible thanks to:
* R `r Citep(bib[["R"]])`
* `r Biocpkg("BiocStyle")` `r Citep(bib[["BiocStyle"]])`
* `r CRANpkg("knitr")` `r Citep(bib[["knitr"]])`
* `r CRANpkg("RefManageR")` `r Citep(bib[["RefManageR"]])`
* `r CRANpkg("rmarkdown")` `r Citep(bib[["rmarkdown"]])`
* `r CRANpkg("sessioninfo")` `r Citep(bib[["sessioninfo"]])`
* `r CRANpkg("testthat")` `r Citep(bib[["testthat"]])`
* `r Biocpkg('AnnotationHub')` `r Citep(bib[['AnnotationHub']])`
* `r CRANpkg('cowplot')` `r Citep(bib[['cowplot']])`
* `r Biocpkg('edgeR')` `r Citep(bib[['edgeR']])`
* `r Biocpkg('ExperimentHub')` `r Citep(bib[['ExperimentHub']])`
* `r CRANpkg("jaffelab")` `r Citep(bib[["jaffelab"]])`
* `r Biocpkg('SummarizedExperiment')` `r Citep(bib[['SummarizedExperiment']])`
* `r CRANpkg('ggplot2')` `r Citep(bib[['ggplot2']])`
* `r CRANpkg('ggrepel')` `r Citep(bib[['ggrepel']])`
* `r Biocpkg('scater')` `r Citep(bib[['scater']])`
* `r CRANpkg('rlang')` `r Citep(bib[['rlang']])`
* `r CRANpkg('gridExtra')` `r Citep(bib[['gridExtra']])`
* `r Biocpkg('variancePartition')` `r Citep(bib[['variancePartition']])`
* `r Biocpkg('limma')` `r Citep(bib[['limma']])`
* `r CRANpkg('VennDiagram')` `r Citep(bib[['VennDiagram']])`
* `r CRANpkg('biomartr')` `r Citep(bib[['biomartr']])`
* `r CRANpkg('Hmisc')` `r Citep(bib[['Hmisc']])`
* `r CRANpkg('R.utils')` `r Citep(bib[['R.utils']])`
* `r Biocpkg('clusterProfiler')` `r Citep(bib[['clusterProfiler']])`
* `r Biocpkg('org.Mm.eg.db')` `r Citep(bib[['org.Mm.eg.db']])`
* `r CRANpkg('pheatmap')` `r Citep(bib[['pheatmap']])`
* `r Biocpkg('GenomicRanges')` `r Citep(bib[['GenomicRanges']])`
* `r Biocpkg('rtracklayer')` `r Citep(bib[['rtracklayer']])`
This package was developed using `r BiocStyle::Biocpkg("biocthis")`.
Date the vignette was generated.
```{r reproduce1, echo=FALSE}
## Date the vignette was generated
Sys.time()
```
Wallclock time spent generating the vignette.
```{r reproduce2, echo=FALSE}
## Processing time in seconds
totalTime <- diff(c(startTime, Sys.time()))
round(totalTime, digits = 3)
```
`R` session information.
```{r reproduce3, echo=FALSE}
## Session info
library("sessioninfo")
options(width = 120)
session_info()
```
# Bibliography
This vignette was generated using `r Biocpkg("BiocStyle")` `r Citep(bib[["BiocStyle"]])`
with `r CRANpkg("knitr")` `r Citep(bib[["knitr"]])` and `r CRANpkg("rmarkdown")` `r Citep(bib[["rmarkdown"]])` running behind the scenes.
Citations made with `r CRANpkg("RefManageR")` `r Citep(bib[["RefManageR"]])`.
```{r vignetteBiblio, results = "asis", echo = FALSE, warning = FALSE, message = FALSE}
## Print bibliography
PrintBibliography(bib, .opts = list(hyperlink = "to.doc", style = "html"))
```